One Health Newsletter
Sickle Cell Disease: A One Health Solution to a Neglected Genetic Disease
Authors
Léon Tshilolo, M.D., Ph.D.
Sickle Cell Disease Research Network in Central Africa (REDAC)
Kinshasa
Democratic Republic of Congo.
Jean-Paul Gonzalez, M.D., Ph.D.
REDAC, Vice President
Georgetown University, Faculty
School of Medicine
Washington D.C., USA
Sickle Cell Disease (SCD) is one of many inherited red blood cell (RBC) diseases of public health significance. By 2050, it is estimated that 14 million children will be born with sickle cell anemia (SCA), the homozygous form of SCD. Over 70% of births will be in Africa.1 SCD causes high mortality and significant morbidity, especially in children. Infection, anemia, and stroke are the most common causes of mortality, while pain is the most frequent cause of morbidity. SCD presents multiple clinical pictures that involve several bodily systems, including cardiovascular system in the broadest sense, from RBCs to micro blood vessels of the bones, immune system and its inflammatory response, musculoskeletal and reticuloendothelial systems, gastrointestinal system, and urogenital system. Ultimately, SCD can affect many organs throughout the body with understandable cognitive and psychological consequences. Fighting the disease engages patients and their families in a constant battle, with increasing success due to neonatal screening for early diagnosis and some emerging, promising therapeutics on the horizon (e.g., marrow transplantation, oxycarbamide treatment, gene therapy). Therefore, SCD is a complex disease that requires an extensive multidisciplinary approach – like the One Health approach – to be understood, controlled, and prevented.
Background
About 10,000 years ago, humans changed from a nomadic to a sedentary way of life, abandoning hunting and gathering in favor of agricultural development. This lifestyle change was a driver of the emergence of zoonotic infectious diseases due to human encroachment into wildlife habitats. One example of this shift is malaria, where the Anopheles spp. mosquito acts as a vector of the Plasmodium parasite. When mosquitoes met the sedentary human, Plasmodium falciparum, which emerged and primarily evolved in West Africa, became responsible for increased disease severity and high mortality associated with malaria in various environments of the tropical zone.
Indeed, Plasmodium's preferred cell target is the RBC, a cell stripped of its nucleus without the ability to produce proteins for immunological defense. The infected person's body compensates for this handicap and resists the Plasmodium parasite by altering hemoglobin, the attacking microbe's primary nutrient. Humans achieved this mechanism of resistance evolutionarily through the adenine-thymine nucleobase substitution through a genetic mutation. This substitution occurs on the 20th nucleotide of the complementary DNA (cDNA), corresponding to codon 7 of the exon of the gene β-globin on chromosome 11, which alters hemoglobin (HgbS). This resulting mutation, called βS, is responsible for SCD, a monogenic hereditary disease common in regions where malaria is prevalent, such as Africa and the Arab-Indian Peninsula. This mutation also confers resistance to severe forms of malaria to heterozygous carriers.2
Unfortunately, this selective advantage is accompanied by the unfavorable consequence; if the mutation is inherited from both parents, each carrying the βS -globin gene (heterozygous or homozygous), it can manifest itself in a homozygous offspring (βS / βS). This primary form of SCD is widespread throughout the world and is responsible for child mortality in Africa and India, where the βS gene's prevalence is high. It is estimated that between 2010 and 2050, there will be a 30% increase in SCD incidence, reaching more than 400,000 new cases per year shared between India and Africa (Figure 1).1
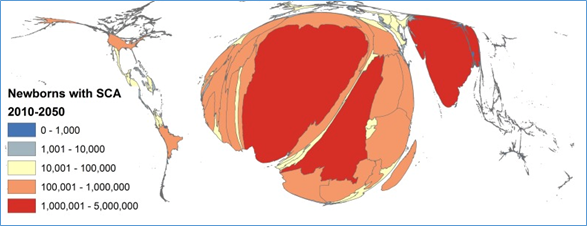
Figure 1: Density-equalizing cartogram of the estimated number of newborns with Sickle Cell Disease (SCD) per country from to 2010 to 2050. (Source: Piel et al. 2013 with the Creative Commons Attribution License).
Disease Pathophysiology
The pathophysiology of this disease is complicated and depends on the geographical environment in which the SCD patient resides. In a hypoxic environment, such as at high altitudes or in a polluted environment with high carbon dioxide levels, the abnormal hemoglobin S (HbS) then precipitates and forms filaments that will alter the cell membrane and causes sickle-shaped deformation of the RBCs (Figure 2). This hypoxic environment of the RBC is undoubtedly harmful to the Plasmodium aggressor, which results in a low level of parasitemia, but is responsible for the severe clinical disease presentation of SCD subjects.3

Figure 2: Red blood cell transformation produced by the sickle cell disease genetic disorder.Legend: A: Blood smear; B: Normal Red Blood Cell; C. Sickle Cell (Source: Open Access of Creative Commons Attribution License)
Thus, RBCs become fragile and undergo hemolysis, which plays a vital role in the complex disease pathophysiology, in which all components of the vascular system (e.g., blood cells, vascular endothelium) are involved.4 However, fortunately, it is based on this complex physiopathology that the up-and-coming therapeutic prospects for SCD are taking shape, ranging from the use of hydroxycarbamide to bone marrow transplantation and gene therapy.
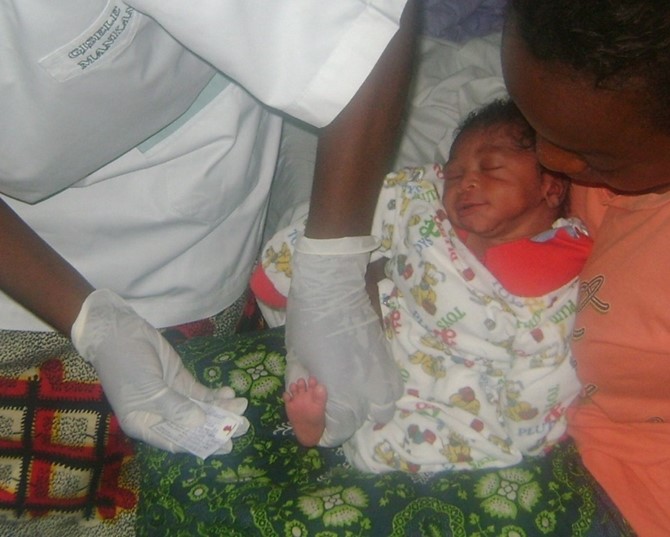
Figure 3: Neonatal screening for sickle cell disease: blood spot collected on filter paper from baby’s heel. (Source: Léon Tshilolo. CEFA Informed consent from the mother.)
SCD is characterized by chronic anemia, often requiring transfusions, frequent painful attacks related to vaso-occlusive crisis, susceptibility to infection, and chronic organ damage.5 The quality of life and life expectancy of the SCD patient depends on the sick person's environment. In Africa, mortality remains very high, and only one child in five will reach the age of five years old, while in Europe and America, more than 90% of children will reach adulthood. This difference depends essentially on the climate, environment, socio-economic conditions, and mainly on the national health policies adopted in these countries. For example, the introduction of neonatal detection of SCD, penicillin-prophylaxis, and adherence to the vaccination calendar are key to a national policy's success to combat SCD, as recommended by the World Health Organization (Figure 3).6 Also, hydroxycarbamide in African settings has proved effective with a significant reduction of complications and mortality in young children.7
These encouraging results should help reduce the burden of SCD in African countries, where patients and families are subject to stigma and discrimination. This negative perception is also a topical issue even in industrialized countries where the quality of care for SCD patients is limited.8 Thus, the fight for SCD patients' well-being involves combating this stigma (Figure 4).
Figure 4: SCD in the Era of the Coronavirus Disease 2019 Pandemic. (Credit: Léon Tshilolo. Monkole Hospital).

The coronavirus disease 2019 (COVID-19) pandemic has shaken up the plans and prospects to combat SCD, starting with the significant congresses planned in 2020, which were canceled or re-organized on a virtual platform (e.g., 4th Global Congress on Sickle Cell Disease in April 2020 http://www.global-scd2020.com/ 8th Sickle Cell Disease Study Network in Central Africa, REDAC Symposium in July 2020, https://redacnetwork.org). In addition to the multiple impacts of the COVID-19 pandemic on people's lives, economies, and resources, this pandemic has not spared those high-risk individuals living with SCD who have a higher mortality rate than the general population.9
Ultimately, the many similarities in clinical expression (e.g., acute respiratory distress, nephropathies, thromboembolic phenomena) and physiopathology (e.g., inflammatory syndrome, cytokine storm) between COVID-19 and SCD has highlighted the high risk presented by subjects carrying the βS mutation. In the United States, this may partially explain the high prevalence of African-American patients suffering from COVID-19.9
In Africa, to our knowledge, there are virtually no published data on SCD patients with COVID-19. The relatively low prevalence and reduced mortality of COVID-19 observed in Africa should lead us to study the impact of the βS gene expression in an environment marked by endemic malaria, numerous viral infections, poverty, promiscuity, lack of adequate medical infrastructure, and lack of social distancing – a context quite different from the industrialized countries.
In conclusion, this example of severe disease shows us the impact it has had in the history of migration, medicine, and human genetics, and the ever-increasing importance of its societal acceptance and care. We can observe SCD as a genetic disease that has competed with an infectious disease in given environments and times through both ancient and present history. Hence, SCD highlights how One Health, trans- and multidisciplinary approaches are indispensable to advancing scientific knowledge.
References
- Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010-2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013;10(7):e1001484.
- Allison AC. Protection afforded by sickle-cell trait against subtertian malareal infection. Br Med J. 1954;1(4857):290-294.
- Elguero E, Délicat-Loembet LM, Rougeron V, et al. Malaria continues to select for sickle cell trait in Central Africa. Proc Natl Acad Sci U S A. 2015;112(22):7051-7054.
- Sundd P, Gladwin MT, Novelli EM. Pathophysiology of sickle cell disease. Annu Rev Pathol. 2019;14:263-292.
- Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017;390(10091):311-323.
- Therrell BL Jr, Lloyd-Puryear MA, Ohene-Frempong K, et al. Empowering newborn screening programs in African countries through establishment of an international collaborative effort. J Community Genet. 2020;11(3):253-268.
- Tshilolo L, Tomlinson G, Williams TN, et al. Hydroxyurea for Children with Sickle Cell Anemia in Sub-Saharan Africa. N Engl J Med. 2019;380(2):121-131.
- Power-Hays A, McGann PT. When actions speak louder than words - Racism and sickle cell disease. N Engl J Med. 2020;383(20):1902-1903.
- McFarling L. Millions of Americans carry the sickle cell trait, many without knowing it. Could they be at risk for severe Covid-19? https://www.statnews.com/2020/09/03/millions-carry-sickle-cell-trait-could-they-be-at-risk-for-severe-covid19/. Accepted November 20, 2020. Updated September 3, 2020.
Contact Us